CRISPR-Cas9是一种基因组编辑工具,它比以前的DNA编辑技术更快、更便宜、更准确,能使遗传学家和医学研究人员通过去除、添加或改变DNA的片段来编辑基因组的一部分,具有广泛的潜在应用。
CRISPR, 1987年日本科学家在大肠杆菌的基因组发现有特别的规律序列,某一小段DNA会一直重复,重复片段之间又有相等长的间隔,此序列称为CRISPR(clustered, regularly interspaced, short palindromic repeats,规律间隔成簇短回文重复序列)。它是存在于细菌中的一种基因,该类基因组中含有曾经攻击过该细菌的病毒的基因片段,细菌透过这些基因片段来侦测并抵抗相同病毒的攻击,并摧毁其DNA。这类基因组是细菌免疫系统的关键组成部分。
Cas9(或“CRISPR相关蛋白9”)是一种酶,它使用CRISPR序列作为引导序列,以识别和切割与CRISPR序列互补的特定DNA链。Cas9酶与CRISPR序列一起构成了CRISPR-Cas9技术的基础,该技术可用于编辑生物体中的基因。这个编辑过程有各种各样的应用,包括基础生物研究、生物技术产品的开发和疾病治疗。值得一提的是,CRISPR-Cas9基因组编辑技术的发展得到了2020年诺贝尔化学奖的认可。
人体有着一套复杂且高效的免疫系统,时刻保护着我们免受病毒和细菌的攻击。但是,对于弱小又无助的原核细胞而言,它们也是急切需要被保护的。为此,经过几亿年的进化,细菌和大部分古细菌衍生出了CRISPR-Cas系统,用于保护它们免受外源DNA和噬菌体的侵染。到目前为止,科学家们发现50%的细菌和超过90%的古细菌均携带有CRISPR-Cas系统。CRISPR是一段高度重复的DNA序列,两端重复序列之间存在着各不相同的spacer序列,表达Cas蛋白家族的基因簇位于CRISPR位点的上游,其表达翻译的几种类型的Cas蛋白作为原核生物免疫应答的效应器发挥着重要作用。
当细菌受到噬菌体的侵染时,被释放到细菌细胞质中的噬菌体基因组的某段DNA序列被识别并整合到CRISPR的spacer区域,随后转录出相应的crRNA前体(pre-crRNA)。crRNA前体经过修饰和加工,生成向导RNA(guide-RNA)。由于gRNA中存在一段来源于噬菌体基因组的序列,因此gRNA可以通过碱基的互补配对原则识别噬菌体的基因组。同时存在于细胞质中的gRNA和Cas蛋白特异性结合,并靶向到噬菌体基因组参与DNA的切割与降解。
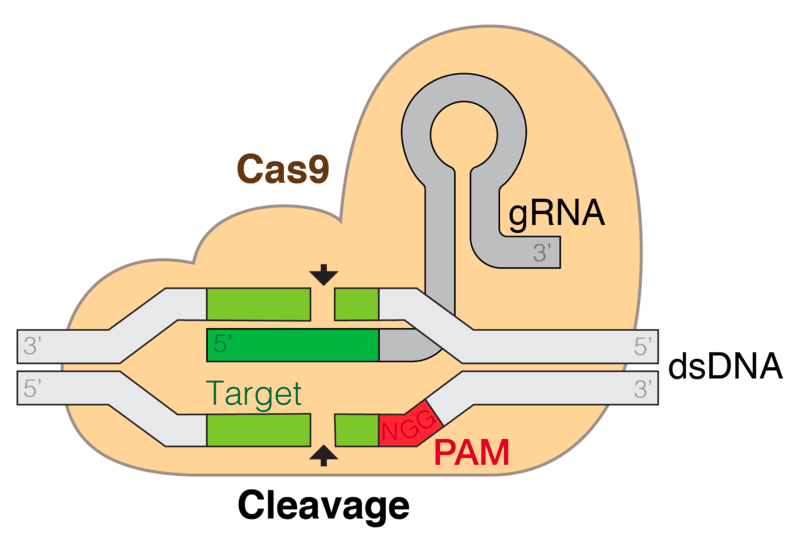
CRISPR-Cas9图示
基因编辑的完成依赖于核酸酶在基因组中特定位置产生双链断裂(DSB)以及细胞本身的修复机制,该过程基本分成三个步骤:靶向目标序列,形成DNA断裂,修复断裂同时引入敲除或敲入。
靶向目标序列:基因编辑的第一步也是最重要的一步就是在基因组上找到需要进行编辑的位置。基因组数据庞大复杂,例如人类的基因组DNA大小达到3GB。在庞大的基因 组中定位到需要剪切的部位需要一个高效、精准的定位方法。
形成DNA断裂:定位到目标基因组位置以后,借助核酸内切酶在DNA双链上形成双链DNA断裂,以此激发细胞本身的DNA修复机制。
修复断裂同时引入敲除或敲入:DNA双链断裂后,会激发细胞本身的DNA修复机制。细胞的DNA损伤修复包含两种主要途径,一种是非同源性末端结合 (Non-homologous End Joining, NHEJ),即易错修复。NHEJ途径会在修复位点引起随机插入或缺失,造成移码突变,使得基因不再表达,由此形成基因敲除。另一种是同源重组介导的修复(Homology directed repair, HDR),即精准修复。HDR途径能 借助外源引入的单链或双链DNA为模板介导基因替换或插入,这种方式可以将一段DNA 序列精准地插入特定的基因组位点,由此完成基因敲入或替换。
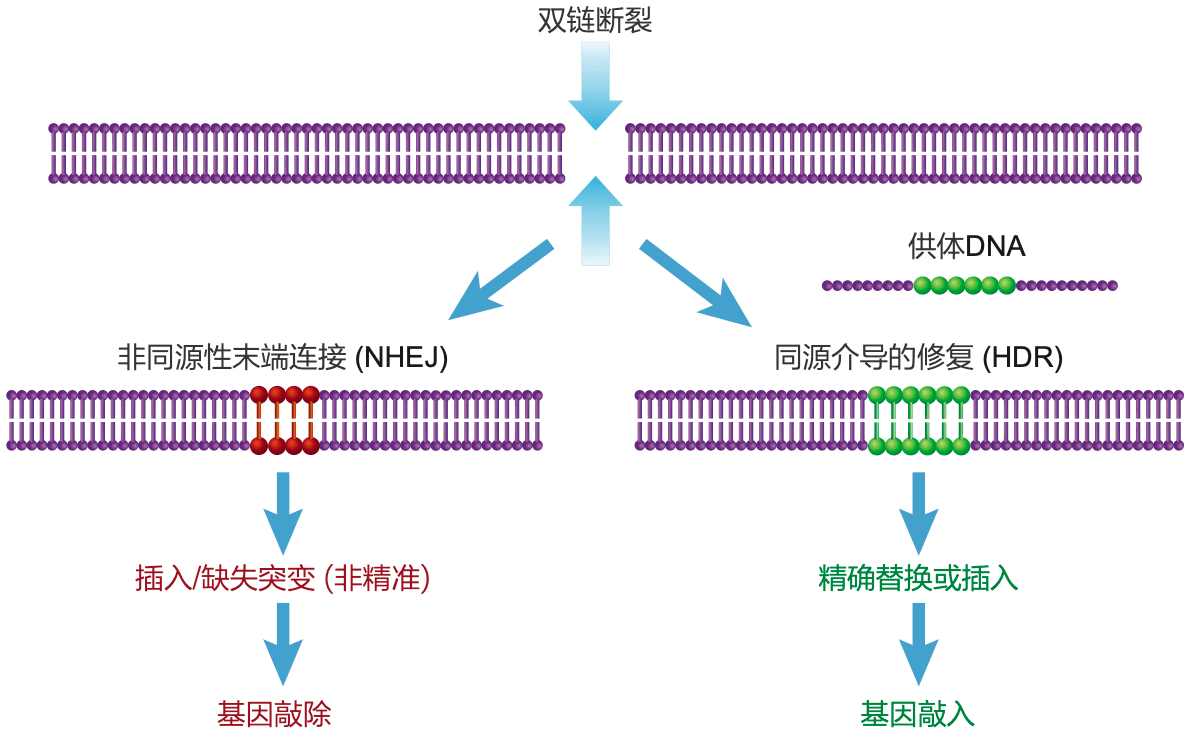
CRISPR/Cas实验原理
CRISPR/Cas系统是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御机制,可对抗入侵的病毒及外源DNA。CRISPR/Cas系统分为两类:第1类包含I、III和IV型,使用多种Cas蛋白的复合体;第2类包含II、V和VI型,只使用一个具有多个结构域的Cas蛋白。第2类CRISPR/Cas系统具有简单、易使用性,更适合基因工程应用,而在第2类CRISPR/Cas系统的不同类型中,II型CRISPR/Cas9是目前研究及应用最为广泛的系统之一。
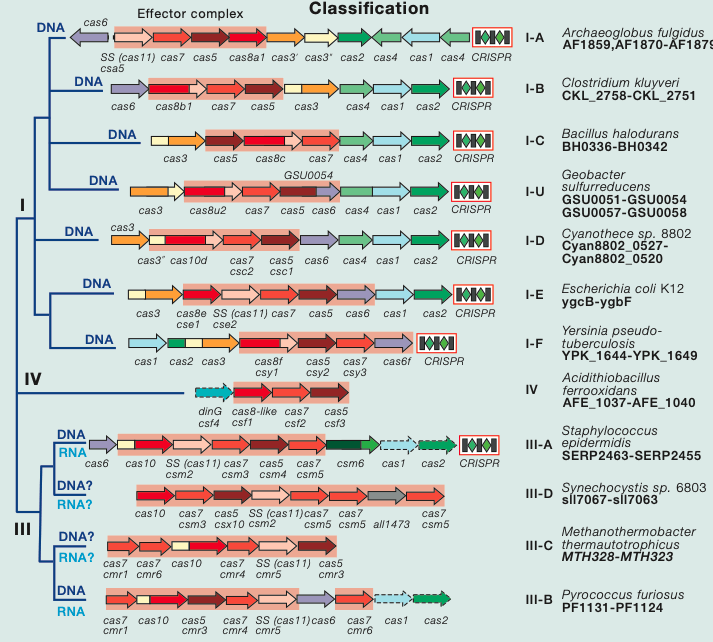
CRISPR-Cas系统分类图示
CRISPR-Cas9其中一种最为重要且广泛的应用便是基因敲除。
那么,基因敲除又是什么呢?
从很久以前,生物学家们就热衷于一种研究策略,那就是功能缺失策略(loss of function)。常规的理解是这样的,当我们想要去研究某个基因在整个基因组中的作用时,只需要通过某种方法将这个基因从基因组中剔除,然后观察去除这个基因之后,这个细胞或生物体会发生怎样的变化,然后根据这些变化去推断这个基因的功能。基因敲除就是用来描述将某个或某些基因从基因组中剔除这件事,而基因敲除技术则是用于完成这件事的方法或技术。
那又该如何实现基因敲除呢?
在实际实验过程中,我们只需要将Cas9蛋白和设计好的gRNA同时“运送”进细胞中即可,接下来的就交给细胞自己去操作了。
那么,对于转导gRNA和Cas蛋白的常见方法主要有四种:
1)RNP法。直接将大量的gRNA和Cas9蛋白,通过电转的方式转染目的细胞
2)病毒法。构建慢病毒(LV)敲除质粒,经病毒包装和纯化后转染细胞,将sgRNA和Cas9蛋白元件整合到细胞基因组中,从而使细胞源源不断地生产sgRNA和Cas9蛋白。
3)常规质粒法。构建常规敲除质粒,通过电转将其导入细胞,短时间内表达gRNA和Cas9蛋白。
4)转座子法(PiggyBac)。将携带有PB转座子和piggyBac转座酶(PBase)的质粒共转染细胞,转座酶促进转座,将高拷贝的Cas9蛋白和gRNA表达元件随机整合到基因组,实现gRNA和Cas9蛋白的持续表达。
实验流程:
设计sgRNA
构建sgRNA-Cas9载体
构建sgRNA-Cas9稳转株
筛细胞克隆并进行qPCR验证
阳性克隆测序,选择敲除純合细胞株
筛选细胞单克隆测序
实验步骤:
设计sgRNA
针对小鼠LIF基因座(Musmusculus,NM_008501)设计了三种sgRNA。进行软件分析以确保sgRNA没有预测的脱靶结合位点。
构建sgRNA-Cas9载体
然后将选择的sgRNA设计克隆到pLenti-U6-sgRNA-SFFV-Cas9-2A-Puro All-in-Onelentivector中(见下图)
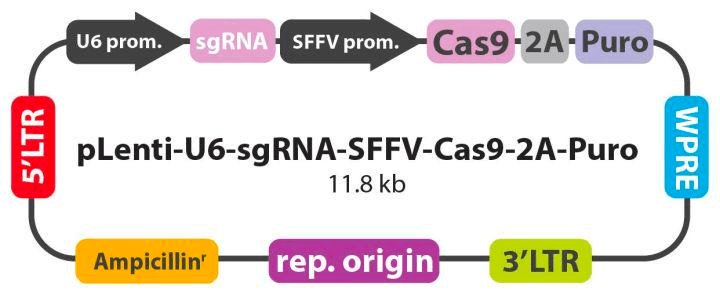
sgRNA-Cas9载体图谱
构建sgRNA-Cas9稳转株
利用慢病毒包装体系构建sgRNA-Cas9稳转株,进行嘌呤筛选
筛细胞克隆并进行PCR验证
嘌呤霉素筛选后分离细胞克隆。提取基因组DNA并进行验证测定目的基因座是否进行编辑
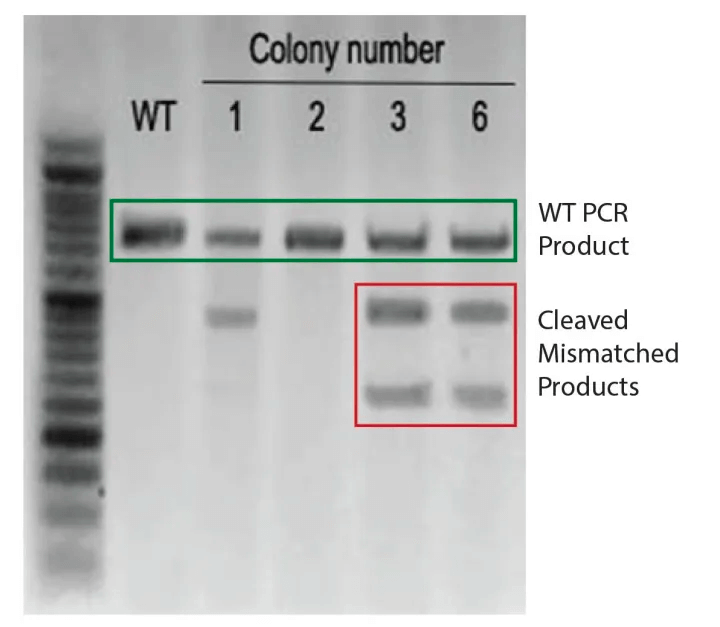
qPCR检测基因组编辑情况
在野生型(WT)测定中的单个条带表示没有发生编辑; 两个较小的条带(总和WT的长度)表示编辑已经发生。克隆 3和6编辑; 克隆2没有编辑; 克隆1是不确定的。
阳性克隆测序,选择突变细胞株
通过Sanger测序进一步分析细胞集落3和6的PCR产物以确定敲除的性质。

细胞基因组测序结果
对于细胞集落3,只检测到一个突变序列,表明这些细胞可能只是杂合敲除。在菌落6中检测到两种不同的突变序列。
筛选细胞选择单克隆纯合突变细胞株
将菌落6连续稀释到96孔板中进行单克隆选择。从这些克隆(即6a,6b..)中提取基因组DNA,进行PCR扩增,克隆和测序。
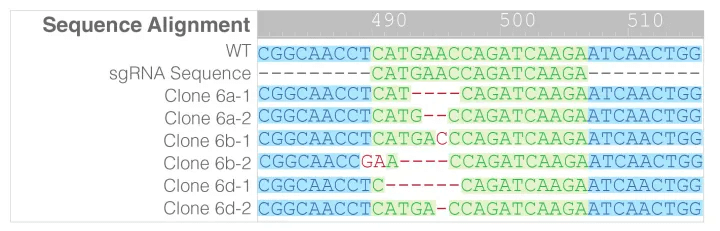
细胞基因组测序结果
测序显示,在两个等位基因中只有克隆6a具有移码突变(图4)。移码突变破坏了开放阅读框架,导致无意义介导的mRNA。
进一步测序,确定纯合突变
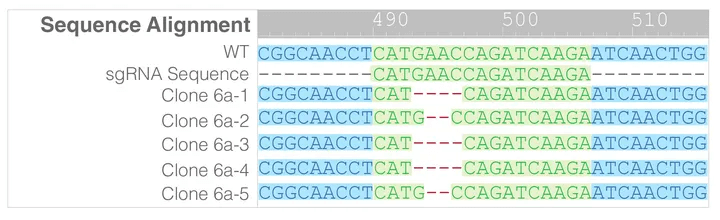
细胞基因组测序结果



我们的地址
杭州术也科技有限公司
浙江省杭州市西湖区西园七路1号绿方科创大厦14层
更多联系方式
座机:(0571) 8876-7926
邮箱:service@vecverse.com