基因克隆
实验背景
基因克隆技术的想法第一次出现在1972年11月在檀香山的一个有关质粒的科学会议上。(质粒是在细菌中发现的DNA环。质粒携带某些对抗生素的耐药性的基因。)一直在研究质粒的史坦利·科恩,对赫伯特·博耶网站上的关于细菌酶切割DNA分子中的特定部分的介绍很感兴趣。在一个深夜游览威基基的一间熟食店两位科学家相遇并谈到将合作领域的科学知识相结合,构思出了一个开创现代生物技术产业的实验。到1973年初,他们推出了一系列的实验,得到方法来选择特定的外源基因在细菌中复制。
实验简介
基因克隆(gene cloning)或分子克隆,又称为重组DNA技术,是应用酶学方法,在体外将不同来源的DNA分子通过酶切、连接等操作重新组装成杂合分子,并使之在适当的宿主细胞中进行扩增,形成大量的子代DNA分子的过程

主要流程及相关原理
一、目的基因的获取
目的基因是指所要研究或应用的基因,也就是将要克隆或表达的基因。获得目的基因是分子克隆过程中最重要的一步。目前用于获得目的基因的方法有几种,如限制性内切酶直接分离法、文库筛选法、体外扩增法和人工合成法等,其中限制性内切酶法直接分离目的基因和多聚酶链式反应(PCR)或逆转录-多聚酶链式反应(RT-PCR)体外扩增目的DNA片段是目前最常用的方法。
(一)采用限制性酶切法直接分离目的基因
- 从原核基因组中制备:原核基因组相对较小,用几种限制性内切酶分别消化原核基因组,或用某种限制性内切酶对所要研究的基因组进行部分消化,可以得到大小不等的各种片段,其中有些片段就会含有目的基因,将这些片段插入载体中进行克隆,经过筛选,可以得到所需的目的基因。
- 从真核基因组中制备:真核基因组比较大,直接用限制性内切酶消化后需要筛选的克隆数太多,不易操作。可以用一种限制性内切酶先消化基因组DNA,产生连续的DNA片段,然后电泳分离这些DNA片段,再用一段特异探针与这些DNA片段进行杂交,在含有特异性模板的区域就会出现杂交带,可以初步鉴定基因组中是否含有目的基因。
- 我们也可以将酶切后的基因组DNA片段全部克隆入适当的载体中,制成基因组文库,再用特异性探针与基因组文库中的不同克隆进行杂交,阳性克隆即表示克隆到的DNA片段含有特异基因序列。将阳性克隆测序,用第一个克隆片段的末端分离下一个克隆片段,然后利用DNA片段间的重叠顺序来鉴定其他克隆。这样一步步走下去,最终可得全部基因序列,这种技术叫做染色体步移(chromosome walking)。
(二)采用PCR或RT-PCR方法制备目的基因
- 根据已发表的基因序列设计并合成引物,采用PCR(以基因组DNA为模板)或RT-PCR(以mRNA为模板)从组织或细胞中获取目的基因片段用于基因操作,这是实验室中最常用的获取已知基因的方法。
- 利用PCR获取目的基因的一大优点就是可以根据需要在引物序列上设计适当的酶切位点、起始密码子或终止密码子等,或通过人为错配改变碱基序列(人工突变)对目的基因进行有限修饰。对于未知基因,可采取构建RT-PCR文库的方法获得未知基因:利用mRNA末端poly A序列尾设计共用引物,将所有mRNA逆转录成cDNA,利用各种工具酶在cDNA末端加尾,然后采用一对共用引物扩增所有cDNA片段,将经PCR扩增后的片段插入到适当载体中,构建成PCR-cDNA文库。这种方法的优点是可以将表达水平很低的mRNA扩增出来,有利于筛选出表达丰度低的基因。由于经过PCR扩增,基因序列的精确性需要采用其他方法进一步确定。
- 还有一个获得目的基因的方法是利用计算机技术进行“计算机克隆”,即利用GenBank中的基因信息,通过软件比较不同种属基因之间的相似性(同源性),利用保守区序列设计引物,再用PCR或RT-PCR从不同种属或不同组织细胞中获取未知的基因片段。这是目前可实现的一种获得新基因的捷径。
(三)基因组文库或cDNA文库的构建和筛选
- 将基因组DNA用限制性内切酶消化后插入到适当载体中,得到含有不同插入片段的克隆载体,这种克隆载体的混合物含有长短不同的基因组片段,这就是基因文库。若将细胞内所有mRNA均逆转录成cDNA,然后将所有cDNA片段克隆到适当的载体中,构建成含有不同cDNA片段的克隆载体混合物,这就是cDNA文库。目前许多组织或细胞的基因组或cDNA文库都可以从商业公司买到。
- 当获得了基因组文库或cDNA文库,可以根据已知的信息合成特异性探针,采用核酸分子杂交的方法从文库中筛选感兴趣的基因片段,这仍是目前获得新基因的一种常用手段。基因组文库或cDNA文库的构建和使用请参见本书相应的章节。
(四)化学合成法制备基因片段
- 采用DNA合成仪,对目的基因进行分段合成,然后进行连接,可以得到所需的目的基因。化学合成法可以改变原始的基因序列,甚至可以合成自然界不存在的序列。在合成过程中可以根据需要改变核苷酸的密码子,如将真核基因序列中不易在E. coli中利用的稀有密码子改成E. coli偏爱的密码子,有利于真核基因在E. coli中的表达。
二、目的基因和载体的连接
获得目的基因后必须将其放在一定的载体内才能在宿主细胞内扩增或表达。目的基因与载体的连接及其后续的转化过程习惯上称为克隆(cloning)。
(一)PCR产物的克隆策略
获得PCR产物通常只是克隆的第一步。无论研究的起始材料是RNA还是DNA,最重要的是要有一种有效的方法对PCR产物进行克隆。目前已建立了多种对PCR产物进行克隆的方法,具体选择哪种方法取决于以下几个因素:PCR产物序列是否已知;载体上有哪些单一的限制性酶切位点可以利用;克隆PCR产物的用途等。
- 限制性内切酶酶切位点添加法:对PCR产物进行克隆的一个基本方法是利用目的片段所特有的限制性内切酶识别位点对产物进行酶切消化。一般是在设计PCR引物时就要考虑到连接方式,直接在引物末端包含与载体相匹配的限制性内切酶位点。
- 设计PCR引物时必须考虑以下几个因素:
①如果PCR产物序列未知,那么在引物上所提供的位点有可能会出现在产物DNA中,使用这些位点进行酶切时将会导致产物内部切割,产生缺失的克隆。
②如果酶的识别位点靠近DNA末端,内切酶对DNA产物的切割效率会降低。可用Klenow酶、T4多聚核苷酸激酶及T4 DNA连接酶等处理PCR产物来克服这一缺陷:首先用激酶对PCR产物进行处理,然后将PCR产物分子上的突出末端用Klenow酶补平,再用T4 DNA连接酶进行连接,形成由多个PCR产物组成的串联体,这种多聚体能被所采用的内切酶有效地切割。
- T/A克隆法:在PCR产物的克隆中,还可以利用含有单个胸腺嘧啶(T)3'突出端的线性化载体与带有单个腺苷酸(A)3'突出端的DNA片段的连接来进行克隆,这种克隆系统被称为T/A克隆。它利用了Taq DNA聚合酶具有延伸酶活性,即以不依赖模板的方式将一个核苷酸添加到已完成延伸的PCR产物的3'末端。对于多数DNA聚合酶,这个添加上去的核苷酸通常是A残基。
- 有几种酶可用来产生3'端带有单个T突出末端的线性化载体,分别是MboII、XcmI和HphI。另外还有两种方法可产生带3'-T突出端的载体,一种是将一个T残基加到经过限制性内切酶线性化处理后产生平端的载体上,或者利用末端转移酶加上一个双脱氧胸腺嘧啶三磷酸(ddTTP);另一种方法则是利用Taq DNA聚合酶具有的延伸酶活性,将一个A残基加到DNA模板的3'端。为了在3'端加上一个T残基,可在高浓度dTTP条件下,将平端载体与Taq DNA聚合酶共同孵育,在其他核苷酸不存在时,载体3'只能加上一个T残基。
(二)外源DNA片段和载体的连接
黏性末端的连接 用一种适当的限制性核酸内切酶将目的基因和载体DNA消化,使它们两端各具有相同的黏性末端,两者的互补末端碱基配对,在DNA连接酶作用下,共价连接成新的DNA分子。
- 单一酶切的黏性末端间的连接:载体分子与外源DNA用同一种限制酶消化后连接有两个缺点:①载体DNA两端的黏性末端可以自身退火,产生载体——载体相连接的假阳性克隆。为了减少和防止这种情况的发生,需用高浓度的外源DNA(插入片段与载体的比率一般为3:1),并用碱性磷酸酶将载体上的5'磷酸基团去掉。②不能定向插入。由于使用同一种内切酶,载体与插入片段的4个末端全为黏性互补顺序,所以外源基因插入可有两个方向。
- 双酶切的黏性末端间的连接:用两种限制性内切酶消化载体和外源DNA片段,因黏性末端不同,载体分子和外源DNA片段只能按一种方向连接,这就是所谓“定向克隆”。
- 平头末端的连接:载体和外源DNA片段的末端是平头也可以连接,但由于平端连接时反应更偏向于载体的自身环化,因此这种方法的效率要比粘端连接低。有几种方法可提高平端连接的连接效率:①在连接反应中使目的基因片段分子数大大过量;②用碱性磷酸酶对载体进行处理,去除载体两端的5'磷酸基团。
三、重组分子的扩增和鉴定
(一)重组DNA分子导入受体细胞
- 目的基因和载体在体外连接形成重组DNA分子后,需要被导入受体细胞中才能进行增殖和(或)表达。接受重组DNA分子的细胞称作受体细胞或宿主细胞。受体细胞分为原核细胞(如大肠杆菌)和真核细胞(如酵母、昆虫细胞及哺乳动物细胞)。原核细胞可作为基因复制扩增的场所,也可作为基因表达的场所;而真核细胞一般只用作基因表达系统。
- 受体细胞是重组基因增殖的场所,所以对受体细胞也应具有几点要求:①容易接纳重组DNA分子;②对载体的复制扩增无严格限制;③不存在特异的能降解外源DNA的内切酶体;④不对外源DNA进行修饰。由于载体的不同,所具备的筛选标志不同,所用的受体细胞也不同,因此可根据所用的载体选择合适的受体细胞。
- 一般将重组DNA分子导入原核细胞的过程称为转化(transformation),而导入真核细胞的过程称为转染(transfection)。
- 未经处理的大肠杆菌很难接受外源重组DNA分子,但经物理或化学方法处理后,细菌对摄取外来DNA分子变得敏感了,这种经处理而易接受外源DNA分子的细胞叫做感受态细胞(competent cell)。
- 将重组DNA分子和感受态大肠杆菌细胞相混合,使重组DNA分子进入大肠杆菌中,就可以实现重组DNA分子的转化。
(二)重组DNA分子的鉴定
重组DNA分子导入受体细胞后是否得到扩增,扩增后的重组DNA分子是否正确,导入的重组DNA分子是否含有正确的插入片段,重组DNA分子能否表达插入的目的基因,一般可以采用以下几种方法对重组DNA分子进行鉴定。
- 抗生素筛选 可根据所选用载体的特性,尤其是抗药基因的存在与否进行初步筛选。例如载体具有抗氨苄青霉素的抗性基因,若转化后的细胞能在含氨苄青霉素的培养基中生长,说明载体DNA被导入到了受体细胞中并且能够扩增繁殖。但这种筛选并不能说明目的基因一定连接到了载体上。但通过抗药基因的失活筛选可以证明有外源基因插入。
- X-gal筛选 有些载体为了方便筛选,根据细菌乳糖操纵子原理,将LacZ基因构建到了载体的多克隆酶切位点处。如果目的基因连接成功,LacZ基因将由于目的基因的插入而失活,不能产生分解乳糖及其类似物的半乳糖苷酶,菌落在含有X-gal的培养基上呈现白色,无目的基因插入的克隆菌落为蓝色。根据这种特性,可以基本判断重组成功与否。
- 酶切电泳鉴定 将重组DNA分子提取出来,用特定内切酶切割重组DNA分子并电泳。如果目的基因被成功地插入到了载体分子中,可以通过目的基因两端的酶切位点将目的基因切割下来,经琼脂糖凝胶电泳即可以判断目的基因的存在与否,这是简便而常用的鉴定方法。
- 序列分析 经过初步鉴定后的重组DNA分子往往需要进行目的基因片段的DNA序列分析,通常采用多克隆酶切位点两端的载体序列作为测序时引物的结合位点,即所谓的通用引物。有关DNA序列分析技术将在有关章节详细叙述。一般需要表达的目的基因都必须经过DNA序列分析予以确认。
- 其他方法 如采用核酸分子杂交或菌落原位杂交鉴定重组DNA分子,或采用蛋白印迹方法直接检测目的蛋白的表达情况。
涉及的主要仪器
- 台式高速离心机
- 外可见光分光光度计
- 高压灭菌锅
- 凝胶成像系统
- PCR仪
- 水平电泳槽
- 电泳仪
- 恒温培养箱
- 恒温摇床
- 恒温水浴箱
- 漩涡振荡器
- 超净工作台
运用
一、疾病模型
很多疾病的致病机制十分复杂,比如癌症,通常涉及多种抑癌基因或致癌基因的遗传改变。因此构建适合的疾病模型对探索疾病的发生和进展以及抗癌药物的筛选有着重要的意义。对于功能未知或者部分未知的基因,研究人员可以通过构建疾病模型从而进一步明确疾病与基因之间的关系。
二、靶向基因治疗
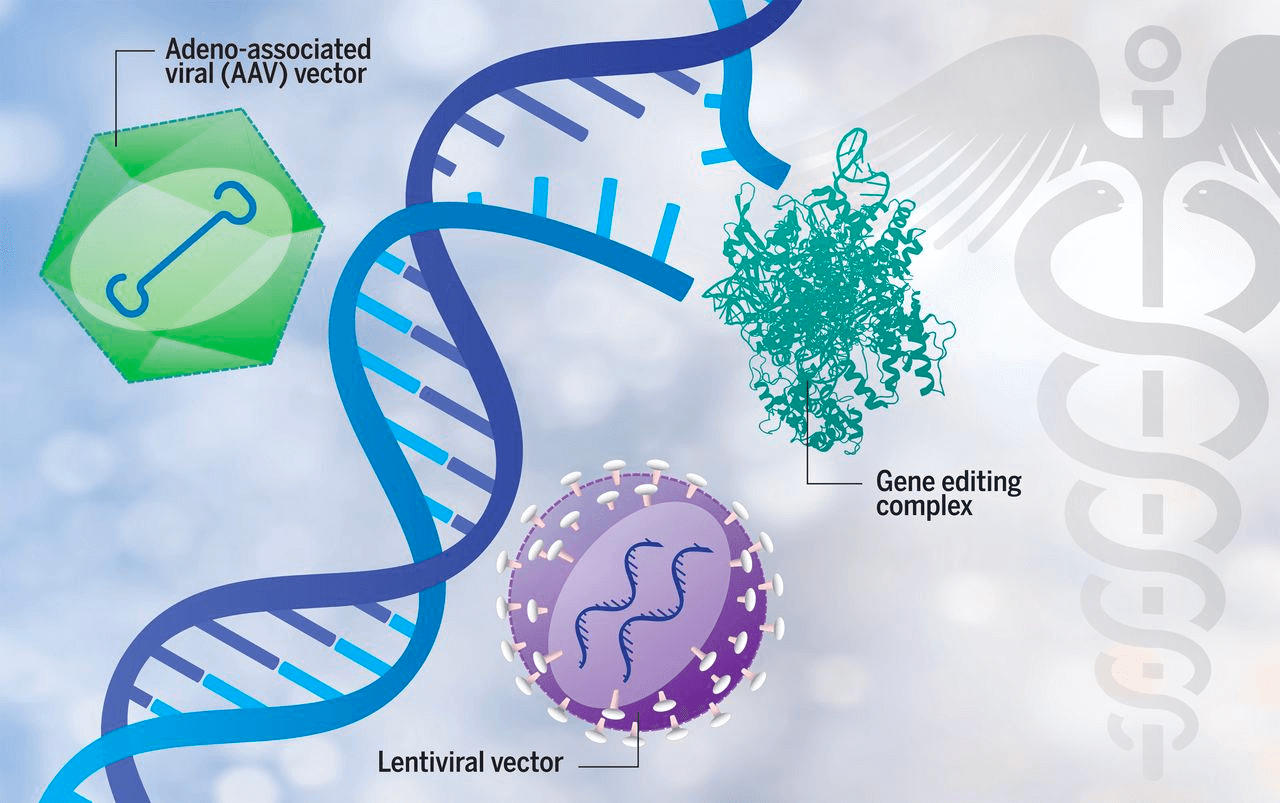
广义上的基因治疗是指在DNA水平上,通过特定的技术手段用正常的基因来替换或者补偿致病基因突变,从而达到治疗目的。常用的基因治疗方法包括用非病毒载体方法、慢病毒载体或腺病毒载体向体内注射正常基因,或者利用近几年崛起的基因编辑技术纠正致病突变等。
三、动植物育种
应用CRISPR/Cas9等技术在农作物或动物基因中进行基因编辑,与传统杂交等育种方法相比,可以精确、快速培育出新品种。在动物品系培养方面,研究者同样可以通过基因编辑技术得到具有优良性状的猪、牛、羊等家畜。
四、微生物设计
基因编辑技术为工业微生物的改造与模式微生物设计提供了高效的工具,为生物燃料、化学品、新材料、医药产品、环境修复微生物等研发提供了新的选择。
五、核酸检测
CRISPR系统具有用一条sgRNA靶向DNA或者RNA的特性,利用该特点研究者开发了一系列的工具用于检测样品中是否存在某种特定的核酸,从而实现即时检测病原体、基因分型和疾病监测等功能。